Caroline A. Chang, MD; Alice B. Gottlieb, MD, PhD; Paul F. Lizzul, MD, PhD, MPH, MBA
CME Released: 09/13/2011; Valid for credit through 09/13/2012
Abstract
Psoriatic arthritis (PsA) is an inflammatory seronegative spondyloarthropathy associated with psoriasis. Although the main assessment measures for PsA are borrowed from the standard criteria used to assess rheumatoid arthritis, a number of new criteria such as the PsAJAI and CPDAI are being developed specifically for PsA. Long-term consequences of untreated PsA include persistent inflammation, progressive joint damage and, in many cases, substantial functional limitations, pain and disability. Moreover, patients with PsA have an increased mortality risk and an increased risk of developing cardiovascular disease and metabolic syndrome. Both GRAPPA and the AAD have developed treatment guidelines, which are discussed here. Psoriasis commonly precedes arthritic symptoms; thus, dermatologists are ideally placed to make the initial diagnosis of PsA and treat it appropriately, affording the opportunity to slow disease progression, improve physical function and enhance quality of life. This Review explores the management of patients with PsA, with a particular emphasis on assessment tools, long-term consequences and treatment issues from the viewpoint of the dermatologist.
Introduction
Current figures suggest that 1-3% of the world population has been diagnosed with psoriasis; psoriatic arthritis (PsA) is an inflammatory, seronegative spondyloarthropathy associated with this condition.
[1] Among patients with psoriasis, the overall prevalence of PsA has been estimated to range from ~11%
[2] to 24%.
[3] In the USA, PsA affects around 520,000 patients, with an overall prevalence of 0.25%.
[2] No gender difference exists in the risk for PsA—although it might occur earlier in women than in men—and PsA can develop at any time during the course of psoriasis.
[4] Symptoms of PsA generally develop after the skin manifestations of psoriasis, with just one in seven patients complaining of arthritis before the onset of cutaneous disease. In many cases, PsA occurs 7-12 years after the initial diagnosis of psoriasis;
[4] for example, skin disease was reported to precede joint disease by a mean of 12 years in 84% of patients with PsA.
[5] Furthermore, the likelihood of developing PsA correlates with duration of clinical psoriasis.
[6,7] Despite these findings, about 14-20% of patients with PsA present with joint manifestations before skin disease.
[8-10] In the absence of a thorough family history, such patients are often classified as having an undifferentiated spondyloarthritis. Given that joint disease can precede cutaneous involvement in some patients with PsA, a diagnosis of 'PsA
sine psoriasis' may occur. Characteristic joint findings in conjunction with HLA-Cw6 positivity and a family history of psoriasis might suggest this diagnosis.
[11]Therefore, the physician must include a complete family history —including family members with psoriasis—in the physical examination of the patient in order to diagnose and treat PsA.
The prevalence of PsA varies with the extent and location of skin involvement. In one study, PsA was observed in 6% of patients with minimal psoriasis, 18% of patients with 3-10% body surface area psoriasis and 56% of patients with >10% body surface area psoriasis.
[2] Importantly, however, the extent of skin disease does not correlate with the severity of joint disease.
[12] Nail dystrophy, scalp lesions, and intergluteal and/or perianal location of psoriatic lesions are associated with an increased likelihood of PsA.
[13] The anatomical relationship between the nail and enthesis is hypothesized to account for the association between PsA and nail disease.
[14]Another study showed a positive association between small joint disease in patients with PsA and onycholysis.
[15]
If left untreated, a substantial number of patients with PsA develop persistent inflammation, progressive joint damage and, in many cases, marked functional limitations, pain and disability.
[16] Rapid diagnosis and therapeutic intervention is, therefore, of paramount consideration. This Review explores the clinical management of PsA from a dermatologist's perspective. Here, we discuss assessment tools, long-term consequences and treatment options for PsA, with a focus on medications familiar to the dermatologist and rheumatologist alike.
Assessment of PsA
The Outcome Measures in Rheumatology (OMERACT) initiative and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) consortium developed a set of six core domains that should be included in all clinical trials for PsA. The fields assessed are peripheral joint activity, skin activity, patient global assessment, pain assessment, physical function and health-related quality of life.
[17]
Quality of Life Tools
The Stanford Health Assessment Questionnaire (HAQ) is easily administered and widely used to predict productivity, morbidity, healthcare utilization, healthcare costs and death associated with rheumatic diseases;
[18] however, this questionnaire has limited relevance for patients with PsA as it does not assess the dermatologic effect of psoriasis. As a consequence, the HAQ-Skin (HAQ-SK) was developed as a PsA-specific tool. HAQ-SK is a short questionnaire that addresses the functional, but not the social or psychological, aspects of psoriasis.
[19] The Psoriatic Arthritis Quality of Life (PsAQoL) tool was later developed to determine the impact of PsA on functional, social, psychological and vocational needs.
[17,20] PsAQoL is an easily administered test in which patients provide 'true' or 'false' answers to a set of 20 questions. Although PsAQoL has demonstrated good reliability, more studies are needed to determine its correlation with treatment effects.
[21]
Treatment Efficacy Tools
Tools to assess treatment efficacy in PsA rely heavily on the standard criteria used to assess rheumatoid arthritis (RA).
ACR20. According to the American College of Rheumatology (ACR) criteria for 20% improvement (ACR20), an improvement of at least 20% in swollen and tender joint count and in three of five other measures (erythrocyte sedimentation rate or C-reactive protein level; physician global assessment of disease activity; patient global assessment of disease activity; patient pain assessment; disability) is indicative of treatment efficacy in patients with PsA.
[22]
DAS28. The Disease Activity Score (DAS)-28 includes the assessment of 28 joints and was originally developed for patients with RA.
[23] DAS28 comprises the following measures: tender joint count; swollen joint count; patient's assessment of pain; patient's and physician's global assessments of disease activity; patient's assessment of physical function; and laboratory evaluation of one acute-phase reactant. However, the joints usually involved in PsA, such as the distal interphalangeal joints of the hands, are not included in the DAS28 assessment. As a consequence, disease activity might be missed with these criteria. In addition, the inclusion of a square root in the DAS28 formula necessitates the use of a calculator in the clinic. Whereas most rheumatologists have access to a hand-held tool that easily calculates the DAS28 score, dermatologists do not have access to or use such a device.
PsARC. The Psoriatic Arthritis Response Criteria (PsARC) were created for a study of sulfasalazine in patients with PsA.
[24] PsARC response is defined by improvement in joint swelling or tenderness in association with improvement in any of four other measures (patient global assessment of articular disease; physician global assessment of articular disease; joint pain or tenderness; joint swelling).
[24,25] Nonetheless, PsARC determines only relative changes from baseline, overestimates the number of responders, has not been formally validated, and is not widely used.
EULAR. The European League Against Rheumatism (EULAR) criteria defines a good response to treatment based on DAS or DAS28 score. Response is measured as improvement in DAS or DAS28 >1.2 and reaching DAS <2.4 or DAS28 <3.2. A study that compared the efficacy of response criteria found that the EULAR criteria were better than ACR20 or PsARC for distinguishing the effects of a TNF inhibitor from placebo in patients with PsA.
[26] However, all three criteria showed improvement in the treatment group and are probably most relevant for PsA patients with active polyarticular disease.
DAPSA. The Disease Activity Index for Psoriatic Arthritis (DAPSA) is derived from the Disease Activity Index for Reactive Arthritis (DAREA) assessment tool; it measures the number of swollen and tender joints, the patient's pain and global assessment, and levels of C-reactive protein. This tool is a useful measure of disease activity for PsA.
[27]
PsAJAI. The PsA Joint Activity Index (PsAJAI) is a simplified score based on ACR30, which involves the weighted sum of 30% improvement in several core measures. This tool is useful as an outcome measure for assessing the response of joint disease in PsA clinical trials.
[28]
CPDAI. The Composite Psoriatic Disease Activity Index (CPDAI) assesses five domains (peripheral arthritis; skin disease; enthesitis; dactylitis; and spinal disease) on a scale of mild (1 point), moderate (2 points) or severe (3 points) in an attempt to create a single composite score for both skin and joint disease. Definitions of what constitutes mild, moderate and severe disease have been defined by the authors and are not universally accepted. CPDAI can distinguish between patients who require a change in treatment regimen and those who do not.
[29]
Joint Progression
The course of PsA varies, with some patients having long-term, mild disease and others experiencing severe, rapid destruction of the joints.
[30] A cohort of 100 patients with PsA was identified and followed prospectively for 5 years.
[31] By study end, 87 patients survived; 18 patients displayed an increased number of joints involved, six had a decrease in joint involvement and one changed from an oligoarticular pattern to predominant spondylitis. Among patients with polyarticular disease at baseline, the majority (73%) had an increase in the number of joints involved, with a median rate of joint progression of 0.42 peripheral joints per year (range 0-7.2).
[31]In a second study, patients were followed prospectively at 6-12 month intervals for >20 years.
[2] Overall, 39% of patients with PsA classified the condition as "a large problem" with everyday life, and an additional 38% rated it "a problem" in everyday life.
[2] These issues stem, in part, from the considerable functional limitations imposed by PsA. A comparison of PsA with RA—which is widely recognized as severely disabling for many patients —showed that, whereas peripheral joint damage is markedly greater in RA than in PsA after equivalent disease duration, function and quality of life scores are nearly identical in the two groups.
[32] These data suggest that PsA should not be viewed as a mild arthropathy. Instead, patients with PsA should be appropriately treated in order to slow progressive structural damage, delay functional limitations and improve quality of life.
Cardiovascular and Metabolic Risk
Patients with PsA have an increased risk of developing cardiovascular disease and metabolic syndrome. In one study, the prevalence of metabolic syndrome in patients with PsA was 58.1% compared with 35.2% of the general population reported by the National Health and Nutrition Examination Survey III.
[33] Metabolic syndrome predisposes patients to diabetes mellitus and atherosclerotic cardiovascular disease. Furthermore, a retrospective database study of patients with PsA in the UK found that cardiovascular disease was the leading cause of death in this group.
[34]
Mortality
RA confers an increased mortality risk, primarily as a result of an increased risk of cardiovascular disease.
[35]However, what is less appreciated is that PsA also confers a considerable incremental increase in mortality risk. Studies conducted in the 1970s and 1980s suggested that patients with PsA had a 1.8-fold increased risk of death compared with the general population.
[36] By contrast, a 1998 study of mortality among 680 patients with PsA found that the risk had dropped to 1.36-fold.
[37] Furthermore, patients with PsA lost 3 years of life on average compared with the general population. Of particular note, radiographic changes observed at presentation are associated with increased mortality risk in patients with PsA.
[37]
Major causes of death among patients with psoriasis include myocardial infarction, respiratory causes, pneumonia, chronic obstructive pulmonary disease and cancer. Patients with PsA have a 5-fold greater risk of death from respiratory disease, compared with a 1.3-fold increased risk of death from cardiovascular disease. The pathophysiological factors underlying the increased risk of pulmonary disease in patients with PsA remain unknown, although data suggest that patients with psoriasis are more likely to smoke than the general population. The link between cardiovascular mortality and PsA might involve metabolic syndrome, a complex constellation of metabolic abnormalities, including diabetes mellitus, hypertension and central obesity, as well as through inflammatory mediators such as TNF.
[36]
Treatment of RA with biologic agents reduces mortality risk and cardiovascular complications,
[38-40] but the effect of such treatment in patients with PsA has not yet been determined. A study of veterans with psoriasis or RA showed that methotrexate decreased the incidence of vascular disease.
[41] Similar treatment of PsA could, therefore, decrease cardiovascular complications of this disease. Long-term, controlled studies with large numbers of patients are needed to demonstrate whether control of inflammation decreases cardiovascular events and mortality in patients with PsA.
Psychological Impairment
Skin and debilitating joint disease both have a profound psychosocial impact on patients with PsA. A positive correlation exists between psoriasis-related disability and the level of alcohol consumption and cigarette smoking and use of psychiatric medications (tranquilizers, sleeping medications and antidepressants).
[42] The severity of both peripheral and axial PsA correlates with poor mental functioning.
[43] In addition, mental health, emotional health and social functioning are decreased in patients with PsA.
[44] Indeed, the reduced quality of life observed with PsA is similar to that seen with other chronic and life-threatening diseases, including cancer, diabetes mellitus and depression.
[45-47] Moreover, 5-20% of patients with psoriasis have contemplated suicide.
[48,49]
General Issues in the Treatment of PsA
Given the many long-term consequences of untreated PsA, early intervention is imperative. Dermatologists and rheumatologists can each play a critical part in the management of patients with PsA by treating upon diagnosis to alleviate signs and symptoms, inhibit structural damage and improve overall quality of life.
[1] However, as cutaneous disease can precede arthritic symptoms by 12 years,
[9]dermatologists may be the first to detect PsA and are, therefore, ideally placed to prevent disability by initiating treatment early.
GRAPPA devised guidelines that categorized each treatment according to categories of evidence (1A-4) and strength of recommendations (grade A to D).
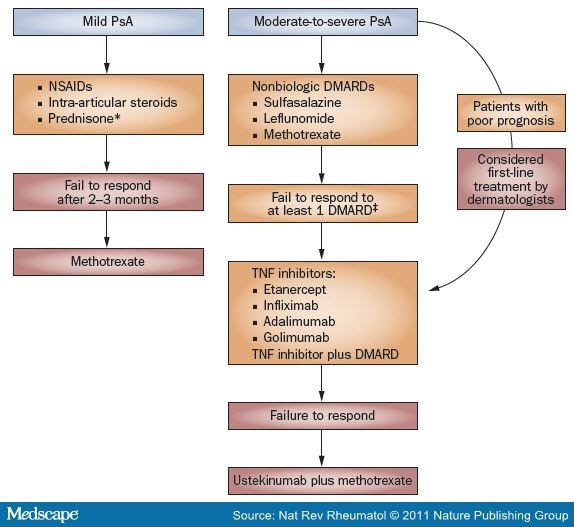
[50] Treatment outcomes emphasize signs and symptoms of disease, quality of life and toxicity. Strength of evidence was delineated for peripheral arthritis, psoriasis, nail involvement, axial disease, enthesitis and dactylitis. A general pathway for the treatment of patients with PsA on the basis of current GRAPPA guidelines is illustrated in Figure 1.
[51]Discussion of treatment is divided into those patients with mild PsA and those with moderate-to-severe disease (
Table 1 ).
Figure 1.
Comparison of GRAPPA51 and AAD52 Treatment Guidelines for Control of Joint Symptoms in PsA. Boxes in orange denote GRAPPA recommendations and those in red denote the AAD guidelines. *Not routinely used by dermatologists secondary to psoriasis flares upon cessation. *Based on EULAR guidelines. §Not FDA-approved for the treatment of PsA. Abbreviations: AAD, American Academy of Dermatology; EULAR, European League Against Rheumatism; GRAPPA, Group for Research and Assessment of Psoriasis and Psoriatic Arthritis; PsA, psoriatic arthritis.
|
The American Academy of Dermatology (AAD) guidelines for PsA differ from those of GRAPPA.
[52] According to the current AAD guidelines, mild PsA should be managed first with NSAIDs alone. If no response is noted after 2-3 months, then methotrexate should be considered. For patients with moderate-to-severe PsA and concurrent psoriasis, treatment with methotrexate, TNF blockade or a combination of these therapies is the first-line option. Ustekinumab with or without methotrexate can be considered as a second-line treatment.
[52] These differences are highlighted in Figure 1.
Drugs used for the treatment of PsA—although generally effective—are subject to a number of warnings, precautions and adverse effects, and must be used with caution. The following discussion should be considered a broad outline of potential safety issues; manufacturer recommendations and the literature must be thoroughly evaluated before initiating therapy with any of these agents.
Treatment of Mild PsA
Mild PsA is generally treated symptomatically with physical therapy, patient education and medication, including NSAIDs and intra-articular injections of corticosteroids.
[1] Neither of these drug classes modifies the course of disease, nor do they inhibit the development of structural joint damage. Dermatologists do not, however, routinely use systemic corticosteroids secondary to the risk of psoriasis flares upon drug cessation.
NSAIDs
Mechanism. NSAIDs reduce inflammation via nonselective inhibition of the cyclooxygenase enzymes COX1 and COX2. This inhibition prevents the formation of prostaglandins and leukotrienes, which contribute to the inflammatory process.
Safety. NSAIDs should not be considered benign drugs. Clinically important NSAID-related events, such as bleeding, result in over 100,000 hospitalizations and 16,500 deaths each year in the USA alone; endoscopic studies indicate that gastric or duodenal ulcers develop in 15-30% of patients who regularly take NSAIDs.
[53]Moreover, these agents are associated with a variable risk of cardiovascular events and might attenuate the efficacy of anti-hypertensive agents, particularly those that act on the renin-angiotensin-aldosterone system.
[54,55]Depending on the patient's level of gastrointestinal risk, nonselective agents may be administered with concomitant proton pump inhibitors. All NSAIDs should be administered with caution to patients with cardiovascular risk factors, and patients should be followed for changes in blood pressure and cardiovascular risk profile. Worsening of psoriasis is rarely observed in patients taking NSAIDs.
[1]
Prednisone
Mechanism. Prednisone is an anti-inflammatory agent that acts through modification of gene transcription via the glucocorticoid receptor. Prednisone is converted in the liver to its active form, prednisolone. The mechanism of blocking inflammation is multifactorial and includes inhibition of COX2, cytokines, cell adhesion molecules, leukocyte infiltration and nitric oxide synthetase.
Safety. Rheumatologists may use low-dose prednisone (5-10 mg daily) for the long-term treatment of PsA.
[1]Adverse effects of this drug depend on dose, duration and frequency of administration. Short courses of prednisone are usually well tolerated; however, long-term, high doses might produce potentially serious adverse effects. Prednisone should be used cautiously in cases of psoriasis because abrupt cessation can precipitate severe psoriasis flares (some requiring hospitalization). As an immunosuppressant, this agent might increase susceptibility to infections such as tuberculosis and decrease response to vaccination. Corticosteroids impair calcium absorption and new bone formation, increasing risk of osteoporosis and subsequent fracture. Guidelines for the prevention of glucocorticoid-induced osteoporosis are available;
[56,57] therapies for preventing bone loss should be continued as long as the patient continues to receive glucocorticoids.
Treatment of Moderate-to-severe PsA
Aggressive therapy with DMARDs is warranted for patients with moderate-to-severe PsA, particularly those individuals with or at risk of progressive structural damage.
Because of lower cost, nonbiologic DMARDs should be considered before biologic agents in most cases. According to the GRAPPA guidelines, TNF inhibitors can be considered a first-line option for patients with a poor prognosis. Indicators of poor prognosis include polyarticular disease, elevated erythrocyte sedimentation rate, previous treatment failures, clinical or radiographic evidence of joint damage, loss of function and decreased quality of life.
[51] A combination of methotrexate and a TNF inhibitor can also be used to treat moderate to severe PsA.
[1]
Nonbiologic DMARDs
Methotrexate. Methotrexate antagonizes the activity of dihydrofolate reductase, which leads to cytoxicity in rapidly dividing cells. Although several nonbiologic DMARDs are available, only methotrexate is currently FDA-approved for the treatment of PsA (despite a lack of evidence for clinical benefit from randomized, controlled clinical trials). By contrast, methotrexate is not approved for the treatment of PsA by the European Medicines Agency (EMA). Data supporting the use of methotrexate as monotherapy in PsA come from two small randomized, placebo-controlled studies.
[58,59] Nonetheless, the low cost of methotrexate usually drives the decision to use it as first-line therapy before TNF inhibitors. Usual dosing for methotrexate in patients with PsA is up to 25 mg per week; concomitant folic acid (1 mg daily) should be given to inhibit methotrexate-induced gastrointestinal effects and reduce the risk of pancytopenia. Methotrexate may be used alone or in combination with biologic agents. Early aggressive therapy with methotrexate at high doses was associated with mild inhibition of joint damage progression in a longitudinal cohort study.
[60] In addition, treatment with methotrexate improved health-related quality of life in patients with PsA.
[61]
The use of methotrexate is contraindicated in pregnant women, alcoholics and noncompliant individuals, as well as in patients with renal impairment, hepatitis or cirrhosis, leukemia or thrombocytopenia.
[62] Drug interactions are frequent and must be accounted for before starting therapy. Bone-marrow suppression, especially with concomitant use of trimethoprim and/or sulfamethoxazole or NSAIDs, is a particularly serious concern. Methotrexate can induce pneumonitis. In addition, it is also a teratogen, an abortifacient, and it decreases sperm count. Indeed, methotrexate is classed as 'pregnancy category X', which implies an increased risk of fetal abnormalities. Methotrexate can cause hepatotoxicity; however, many rheumatologists believe that liver biopsy is not necessary in healthy patients. By contrast, dermatology guidelines divide patients with psoriasis into low-risk and high-risk categories. Individuals at low risk of liver injury should follow the current ACR criteria for methotrexate monitoring: liver biopsy should be considered after a total cumulative dose of 3.5-4.0 g. However, in patients with risk factors for liver disease, such as obesity or diabetes mellitus, or in those with pre-existing liver disease, the dermatology literature recommends monitoring and a liver biopsy after a 1.5 g cumulative dose of methotrexate.
[63]
Sulfasalazine. In a small randomized, controlled trial, administration of 2 g sulfasalazine daily provided a modest benefit in terms of PsARC criteria (57.8% versus 44.6% with placebo).
[24] However, this drug is not yet approved by the FDA or EMA for treatment of PsA.
Sulfasalazine is contraindicated in patients with porphyria, intestinal or urinary obstruction and hyper-sensitivity to sulfasalazine, its metabolites or salicylates.
[64] Sulfasalazine should be used with caution in patients with severe allergies or bronchial asthma, hepatic or renal damage or blood dyscrasias. Adequate fluid intake must be maintained to prevent crystalluria and stone formation. Gastrointestinal upset is a frequent occurrence and can be a limiting factor in dosing. Complete blood analysis, including differential white cell count and liver function tests, should be performed before starting sulfasalazine and monthly for the first 3 months of treatment. Thereafter, monitoring should be performed every 6 months. Urinalysis and an assessment of renal function should also be done periodically during treatment. Sulfasalazine is classified as 'pregnancy category B, which implies low risk of fetal abnormalities.
Leflunomide. Leflunomide is an immunomodulatory drug that inhibits dihydroorotate dehydrogenase, affecting
de novo synthesis of uridine monophosphate. In a small study, a benefit of leflunomide was demonstrated in patients with PsA, with a PsARC response observed in 58.9% of treated patients compared with 29.7% of those who received placebo at an oral dosing regimen of 100 mg per day for 3 days followed by 20 mg per day thereafter.
[65]Leflunomide is approved in Europe (but not in the USA) for treatment of PsA.
Leflunomide is a pregnancy category X drug and is absolutely contraindicated in pregnant women and in patients who plan to become pregnant within 2 years of stopping the drug, unless a regimen of cholestyramine (8 g three times daily for 11 days) is completed and drug concentrations of leflunomide in the blood are below 0.02 mg/l.
[66]Caution should be used in patients with chronic renal insufficiency and those with hepatic insufficiency. Rare cases of severe liver injury, including fatalities, have been reported during treatment with leflunomide.
[66] This drug is not recommended in patients with severe immunodeficiency, bone marrow dysplasia or severe, uncontrolled infections.
[66] Platelet, white blood cell count, hemoglobin or hematocrit and liver function tests should be conducted in accordance with the recommendations of the manufacturer.
[66]
Established Biologic DMARDs
Four TNF inhibitors—adalimumab, etanercept, infliximab and golimumab—are currently the only FDA-approved biologic agents for the treatment of PsA. Administration of adalimumab, infliximab, etanercept or golimumab enables patients to achieve an ACR20 response (
Table 2 ). These drugs also exhibit efficacy in terms of improvements in Psoriasis Area and Severity Index (PASI), which is the gold standard tool for assessing the severity of psoriasis. PASI places an objective number on a patient's psoriasis by evaluating the features of the psoriatic plaque (redness, scaling and thickness) and the extent of involvement. In clinical studies, PASI scores are listed as the percentage of patients who experienced a reduction in their baseline score; for example, a PASI 75 of 50% implies that 50% of study patients achieved a 75% reduction in PASI score from their baseline. When used as monotherapy in patients with psoriasis, both adalimumab and infliximab produce PASI 75 response rates in 71-80% of patients, whereas the response rate with etanercept is somewhat lower (
Table 2 ). For patients with PsA receiving combination therapy, 58-68% of patients receiving either adalimumab, infliximab or golimumab achieved PASI 75, whereas etanercept exhibited a lower response (
Table 2 ). A summary of administration recommendations for approved biologic DMARDs can be found in
Box 1 .
Other biologic agents for the treatment of PsA are also in the pipeline. For example, ustekinumab, a human monoclonal antibody directed against IL-12 and IL-23, is approved for the treatment of psoriasis but also shows promise in PsA.
[67]
TNF Inhibitors: General Efficacy Considerations. Adalimumab, etanercept and infliximab are FDA-approved and EMA-approved to control signs and symptoms, inhibit radiographic progression and improve quality of life in patients with PsA. Golimumab is currently FDA-approved and EMA-approved for only control of signs and symptoms, in PsA but not psoriasis.
Loss of efficacy over time has been observed with all TNF inhibitors, a phenomenon potentially related to the development of antibodies against the biologic drug. Some dermatologists, therefore, prescribe concomitant low-dose methotrexate, with the goal of limiting antibody formation and maintaining efficacy over time.
[68] Limited data in psoriasis,
[69] PsA,
[70] RA
[71] and inflammatory bowel disease
[72] suggest that switching from one TNF inhibitor to another following therapeutic failure or intolerance to the first agent can result in substantial clinical efficacy. Moreover, adding low-dose methotrexate to biologic treatments might reduce the immunogenicity associated with monoclonal antibody TNF inhibitors.
[73,74]
Adalimumab. Adalimumab is derived from a fully human anti-TNF monoclonal antibody that is effective in a number of inflammatory conditions, including psoriasis and RA, when used alone or in combination with methotrexate.
[75] Adalimumab currently carries a guideline 1A recommendation (highest strength of recommendation, high level of evidence) for the treatment of PsA.
[1]
The efficacy of adalimumab in patients with PsA was initially assessed in a double-blind, randomized, controlled trial that enrolled adults with moderate to severe disease (defined as at least three swollen joints and three tender or painful joints), in association with active psoriatic skin lesions or a documented history of psoriasis.
[76]Participants received either adalimumab (40 mg given subcutaneously every other week for 24 weeks) or placebo. Patients who received adalimumab had an appreciably higher ACR20 response rate than those who received placebo at all time points assessed (58% versus 14% at week 12). ACR50 and ACR70 response rates were also markedly higher among patients who received adalimumab.
[76] An open-label extension of this trial demonstrated that efficacy is maintained, with ACR20, ACR50, and ACR70 response rates of 56%, 44%, and 30%, respectively, at week 24. Sustained improvements in the Disability Index of the HAQ, and stabilization in changes in radiographic parameters were also observed with adalimumab;
[77] inhibition of radiographic progression and improvements in joint disease were maintained in most patients at 2 years.
[78]
Etanercept. Etanercept is a fusion protein engineered to link soluble TNF receptor to the Fc component of human IgG that is administered subcutaneously, either as a single 50 mg weekly dose or as two 25 mg doses given on the same day or 3-4 days apart.
[79] This drug is approved for the treatment of both psoriasis and PsA, and carries a 1A guideline recommendation for the treatment of PsA.
[1]
Etanercept was evaluated in a double-blind, randomized, controlled trial of 205 patients with PsA who received placebo or 25 mg etanercept for 24 weeks, with an optional 24-week open-label extension.
[80] Compared with placebo, etanercept was associated with a significant reduction in the signs and symptoms of PsA. At 12 weeks, ACR20 criteria were met by 59% of the etanercept groups, compared with 15% of the placebo group, with efficacy maintained at 24 and 48 weeks.
[80] Radiographic disease progression was inhibited among patients who received etanercept, with an annualized rate of change in the modified total Sharp score of -0.03 units, compared with +1.00 units in the placebo group. The benefit of etanercept was maintained at 2 years.
The Experience Diagnosing, Understanding Care, and Treatment with Etanercept (EDUCATE) trial showed that 50 mg subcutaneous etanercept once a week improved patient global assessment of joint pain and joint disease at weeks 12 and 24 in patients with active PsA treated in a dermatology clinic.
[9] In addition, PsA was newly diagnosed in 23% of psoriasis patients seen in dermatology clinics.
[5] Moreover, in the PRESTA (Psoriasis Randomized Etanerept Study in Subjects with Psoriatic Arthritis) study, three in four patients showed joint improvement, as measured by PsARC, at 12 weeks that was maintained at 24 weeks in both arms of the trial, which compared twice-weekly with once-weekly dosing of etanercept.
[81]
Infliximab. Infliximab is a chimeric (mouse-human) monoclonal antibody directed against TNF, and has a 1A guideline recommendation for the treatment of PsA.
[1] Unlike adalimumab and etanercept, which are subcutaneously administered, 5 mg/kg infliximab is infused over a 2-h period at treatment start and after 2 and 6 weeks, and every 8 weeks thereafter.
[82]
The efficacy of infliximab in treating PsA has been evaluated in multiple trials. In the IMPACT 2 (Infliximab Multinational Psoriatic Arthritis Controlled Trial) study, 200 patients with PsA, who were unresponsive to previous treatment, were randomly allocated to receive infliximab or placebo.
[83] A week 14 ACR20 response was achieved by 58% of the infliximab-treated patients and 11% of patients who received placebo; this response was maintained throughout the 24-week duration of the trial. Similar patterns were observed for ACR50 and ACR70 response. When compared with the placebo group, fewer patients in the infliximab group had dactylis (18% versus 30%;
P = 0.025) or active enthesopathy (22% versus 34%,
P = 0.016). Of note, 1-year analyses of the results of the IMPACT and IMPACT 2 trials showed that infliximab markedly inhibited radiographic progression of PsA.
[84,85]
Golimumab. Golimumab, the newest entrant in the anti-TNF class, is a human monoclonal antibody with a half-life of around 2 weeks, which allows it to be administered less frequently than currently approved drugs for PsA.
[86] A 2009 study evaluated golimumab in the treatment of PsA. Adults with active psoriasis plus at least three swollen joints and three tender joints were randomly allocated to receive placebo, 50 mg golimumab or 100 mg golimumab every 4 weeks for 20 weeks.
[86] At week 14, 48% of all patients who received golimumab (51% of those administered the 50 mg dose and 45% of those receiving the 100 mg dose) achieved an ACR20 response, compared with only 9% of patients who received placebo; similar patterns were observed for ACR50 and ACR70 responses. Marked improvements in quality of life were also observed.
TNF Inhibitors: General Safety Considerations. General safety recommendations for TNF inhibitors are briefly summarized in
Box 2 .
[68] Although the occurrence is infrequent, TNF inhibitors are associated with an increased risk of bacterial, viral, invasive fungal and mycobacterial infections, making careful monitoring and early evaluation critical.
[68] These agents should be avoided in patients with chronic, serious or recurring infections; notably, reactivation of hepatitis B virus infection has been observed in some patients taking TNF inhibitors. Risk of peripheral and central demyelinating disease, including multiple sclerosis, might be increased in patients receiving TNF inhibitors; these agents have been shown to worsen existing demyelinating disease in some cases. Moreover, new-onset psoriasis has been reported in patients administered TNF inhibitors for other diseases.
[87]
Increased incidence of lymphomas and other malignancies have also been reported among patients taking TNF inhibitors. About half of the malignancies reported in children were lymphomas.
[75,79,82] In addition, there are rare reports of aggressive hepatosplenic T-cell lymphoma in adolescents being treated with TNF inhibitors (concomitant with other immunosuppressants) for Crohn disease or ulcerative colitis.
[75,82] Periodic skin examinations are recommended for patients taking TNF inhibitors, as melanoma and nonmelanoma skin cancers have been reported. Merkel cell carcinoma has infrequently been reported in patients treated with etanercept.
[79]Thus, the decision to use a TNF inhibitor should be carefully considered in children and patients with a history of malignancy.
The use of TNF inhibitors should be avoided, if possible, in patients with severe congestive heart failure (New York Heart Association class III or IV), and withdrawn in patients with milder congestive heart failure who exhibit new symptoms or worsening of disease.
[68] Patients administered TNF inhibitors might develop increased levels of circulating antinuclear antibodies, potentially resulting in a drug-induced lupus-like syndrome. Elevated levels of liver enzymes have been observed in patients receiving infliximab, but not adalimumab or etanercept. TNF inhibitors are pregnancy category B.
Novel Biologic DMARDs
Ustekinumab. Ustekinumab, a human monoclonal antibody against the p40 subunits of IL-12 and IL-23, represents an entirely new therapeutic approach for the treatment of PsA that could be useful in patients who do not respond to standard pharmacotherapy. This drug is FDA-approved for use in patients with psoriasis; however, it is not yet approved for the treatment of PsA. Ustekinumab is administered subcutaneously and under the supervision of a physician.
In a double-blind, randomized, controlled study, patients with active PsA received 63 or 90 mg ustekinumab every week for 4 weeks, followed by placebo at weeks 12 and 16; a second group of patients received placebo for 4 weeks and 63 mg ustekinumab at weeks 12 and 16.
[67] The primary end point was ACR20 response at week 12; however, clinical response was followed to week 36. ACR responses were achieved more frequently among patients who received ustekinumab than among those who received placebo during the first 12 weeks of the trial: ACR20 (42% versus 14%;
P = 0.0002), ACR50 (25% versus 7%;
P = 0.0038) and ACR70 (11% versus 0%;
P = 0.0055). Roughly three in four patients initially treated with ustekinumab maintained ACR20 responses at week 36 with no additional treatment received after week 3. Moreover, fewer patients who received ustekinumab experienced enthesopathy (23% versus 42% with placebo;
P = 0.0163).
Some important differences exist in the patient populations enrolled in the TNF blocker and ustekinumab trials. The ustekinumab trial recruited patients from dermatology practices and participants had to have active plaque psoriasis. Up to 31% of patients were previously exposed to biologic agents, only 20% of patients were concomitantly taking methotrexate, 51% of patients received NSAIDS and no patients were concurrently taking oral corticosteroids. Finally, patients had only moderately elevated acute-phase reactant response, as measured by C-reactive protein levels. There were fewer patients with polyarticular joint disease in this study compared with those studying infliximab, adalimumab and etanercept.
Ustekinumab might increase the risk of new infections and reactivation of latent infections.
[88] As such, it should not be given in patients with any clinically important active infection. Of note, patients genetically deficient in IL-12 and/or IL-23 are particularly vulnerable to disseminated infections from mycobacteria and Bacillus Calmette-Guerin vaccinations, although whether ustekinumab has a similar effect is unknown. Nonetheless, patients should be evaluated for tuberculosis infection before initiating treatment and ustekinumab should not be administered with a live vaccine or to patients in close contact with individuals who have been administered a live vaccine. As with other immunosuppressive agents, ustekinumab might increase malignancy risk.
Abatacept. Abatacept is a fully human CTLA-4-IgG1 fusion protein that selectively binds to CD80 or CD86, which blocks T-cell interaction with CD28, the co-stimulatory signal for T-cell activation. Abatacept is not currently approved by the FDA or EMA for the treatment of PsA, although some promising results have been reported. In a phase II trial in patients with PsA and psoriasis who had failed previous therapy with a DMARD, participants were randomly allocated to receive placebo or one of three different doses of abatacept on days 1, 15, and 29 of the study, and then every 28 days thereafter.
[89] ACR20 responses were achieved in 33%, 48%, and 42% of the treatment groups (3 mg/kg, 10 mg/kg or 30 mg/kg intravenous for two doses followed by 10 mg/kg intravenous, respectively) compared with 19% in the placebo group. Further studies are needed, however, to fully assess the efficacy of this drug in the treatment of PsA.
Abatacept inhibits T-cell activation and, therefore, suppresses host response to infections and malignancies. Abatacept increases risk of infection, particularly among patients receiving concomitant TNF inhibitor therapy. Patients with a history of recurrent infections or underlying conditions that might predispose to infection should use caution when taking abatacept. Screening for tuberculosis and hepatitis B should be performed before initiating treatment. In addition, live vaccines should not be given while a patient is taking abatacept. Increased risk of lung cancer and lymphoma has been observed in patients treated with abatacept, and respiratory disorders were seen more frequently in patients with chronic obstructive pulmonary disease. Infusion-related events include dizziness, headache and hypertension. Abatacept is a 'pregnancy category C' drug, which implies that risk to the fetus is unknown or unclear.
Conclusions
A number of treatments are available for PsA, ranging from agents that primarily provide symptomatic relief (NSAIDs and intra-articular corticosteroids) to non-biologic and biologic DMARDs (methotrexate, TNF inhibitors, ustekinumab and abatacept). Limited clinical trial evidence supports the use of methotrexate in patients with PsA, and there are no compelling data to demonstrate that it has a disease-modifying effect in PsA. Nevertheless, methotrexate is used before biologic agents due to its lower cost. In contrast to methotrexate, substantial evidence exists that TNF inhibitors are effective in slowing progressive joint damage specifically in patients with PsA. Novel agents (as yet not approved for PsA), such as ustekinumab and abatacept, may broaden the spectrum of treatments available to patients with PsA.
When left untreated, a substantial proportion of patients with PsA will develop persistent inflammation, which leads to progressive joint damage and—ultimately—to pain, severe functional limitations and disability. Given the long-term functional consequences of unchecked PsA—and the potential benefits of early treatment— dermatologists are strongly encouraged to assess patients for the signs and symptoms of PsA at each visit. Moreover, rheumatologists should be included early in the assessment of patients thought to have PsA. Early detection and treatment can prevent the disability that is associated with untreated PsA.
Key Points
- Dermatologists are ideally placed to diagnose and treat psoriatic arthritis (PsA), as most patients present with skin symptoms before onset of arthritis
- PsA is a potentially debilitating and destructive disease that should be treated as soon as possible after diagnosis to prevent irreversible damage
- Patients with mild PsA can be managed with NSAIDs; however, systemic corticosteroids should be used cautiously in such cases
- Moderate-to-severe cases of PsA can be managed by DMARDs or TNF inhibitors, administered either alone or in combination
- Promising new drugs for the treatment of PsA include ustekinumab and abatacept
References
- Gottlieb, A. et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 2. Psoriatic arthritis: overview and guidelines of care for treatment with an emphasis on the biologics. J. Am. Acad. Dermatol. 58, 851-864 (2008).
- Gelfand, J. M. et al. Epidemiology of psoriatic arthritis in the population of the United States. J. Am. Acad. Dermatol. 53, 573 (2005).
- Prey, S. et al. Assessment of risk of psoriatic arthritis in patients with plaque psoriasis: a systematic review of the literature. J. Eur. Acad. Dermatol. Venereol. 24 (Suppl. 2), 31-35 (2010).
- Qureshi, A. A., Husni, M. E. & Mody, E. Psoriatic arthritis and psoriasis: need for a multidisciplinary approach.Semin. Cutan. Med.Surg. 24, 46-51 (2005).
- Gottlieb, A. B. et al. Clinical characteristics of psoriatic arthritis and psoriasis in dermatologists' offices. J. Dermatolog. Treat. 17, 279-287 (2006).
- Christophers, E. et al. The risk of psoriatic arthritis remains constant following initial diagnosis of psoriasis among patients seen in European dermatology clinics. J. Eur. Acad. Dermatol. Venereol. 24, 548-554 (2010).
- Carneiro, C., Veradino, G., Ramos-e-Silva, M. & Carneiro, S. Psoriasis: Correlation between joint and ungual involvement, extension and duration of cutaneous disease, and quality of life. J. Am. Acad. Dermatol. 62 (Suppl. 1), AB139 (2010).
- Olivieri, I., Padula, A., D'Angelo, S. & Cutro, M. S. Psoriatic arthritis sine psoriasis. J. Rheumatol.Suppl. 83, 28-29 (2009).
- Gottlieb, A. B. et al. Use of etanercept for psoriatic arthritis in the dermatology clinic: the Experience Diagnosing, Understanding Care, and Treatment with Etanercept (EDUCATE) study. J. Dermatolog. Treat. 17,343-352 (2006).
- Gladman, D. D. & Chandran, V. Observational cohort studies: lessons learnt from the University of Toronto Psoriatic Arthritis Program. Rheumatology (Oxford) 50, 25-31 (2011).
- Scarpa, R. et al. Clinical and genetic aspects of psoriatic arthritis "sine psoriasis". J. Rheumatol. 30, 2638-2640 (2003).
- Wittkowski, K. M. et al. Clinical symptoms of skin, nails, and joints manifest independently in patients with concomitant psoriasis and psoriatic arthritis. PLoS ONE 6, e20279 (2011).
- Wilson, F. C. et al. Incidence and clinical predictors of psoriatic arthritis in patients with psoriasis: a population-based study. ArthritisRheum. 61, 233-239 (2009).
- McGonagle, D. Enthesitis: an autoinflammatory lesion linking nail and joint involvement in psoriatic disease. J. Eur. Acad. Dermatol.Venereol. 23 (Suppl. 1), 9-13 (2009).
- Love, T. J., Gudjonsson, J. E., Valdimarsson, H. & Gudbjornsson, B. Small joint involvement in psoriatic arthritis is associated with onycholysis: the Reykjavik Psoriatic Arthritis Study. Scand. J. Rheumatol. 39, 299-302 (2010).
- Gladman, D. D., Antoni, C., Mease, P., Clegg, D. O. & Nash, P. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann. Rheum. Dis. 64 (Suppl. 2), ii14-ii17 (2005).
- Gladman, D. D. et al. Consensus on a core set of domains for psoriatic arthritis. J. Rheumatol. 34, 1167-1170 (2007).
- Bruce, B. & Fries, J. F. The Stanford Health Assessment Questionnaire: a review of its history, issues, progress, and documentation. J. Rheumatol. 30, 167-178 (2003).
- Husted, J. A., Gladman, D. D., Long, J. A. & Farewell, V. T. A modified version of the Health Assessment Questionnaire (HAQ) for psoriatic arthritis. Clin. Exp. Rheumatol. 13, 439-443 (1995).
- McKenna, S. P. et al. Development of the PsAQoL: a quality of life instrument specific to psoriatic arthritis. Ann. Rheum. Dis. 63, 162-169 (2004).
- Mease, P. J. Assessing the impact of psoriatic arthritis on patient function and quality of life: lessons learned from other rheumatologic conditions. Semin. Arthritis Rheum. 38, 320-335 (2009).
- Felson, D. T. et al. The American College of Rheumatology preliminary core set of disease activity measures for rheumatoid arthritis clinical trials. The Committee on Outcome Measures in Rheumatoid Arthritis Clinical Trials.ArthritisRheum. 36, 729-740 (1993).
- Gladman, D. D. et al. Outcome measures in psoriatic arthritis. J. Rheumatol. 34, 1159-1166 (2007) .
- Clegg, D. O. et al. Comparison of sulfasalazine and placebo in the treatment of psoriatic arthritis. A Department of Veterans Affairs Cooperative Study. Arthritis Rheum. 39, 2013-2020 (1996).
- Mease, P. J. et al. Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised trial. Lancet356, 385-390 (2000).
- Fransen, J. et al. Performance of response criteria for assessing peripheral arthritis in patients with psoriatic arthritis: analysis of data from randomised controlled trials of two tumour necrosis factor inhibitors. Ann. Rheum. Dis. 65, 1373-1378 (2006).
- Schoels, M. et al. Application of the DAREA/ DAPSA score for assessment of disease activity in psoriatic arthritis. Ann. Rheum. Dis. 69, 1441-1447 (2010).
- Gladman, D. D., Tom, B. D., Mease, P. J. & Farewell, V. T. Informing response criteria for psoriatic arthritis. I: discrimination models based on data from 3 anti-tumor necrosis factor randomized studies. J. Rheumatol. 37,1892-1897 (2010).
- Mumtaz, A. et al. Development of a preliminary composite disease activity index in psoriatic arthritis. Ann. Rheum. Dis. 70, 272-277 (2011).
- Lebwohl, M. Psoriasis. Lancet 361, 1197-1204 (2003).
- McHugh, N. J., Balachrishnan, C. & Jones, S. M. Progression of peripheral joint disease in psoriatic arthritis: a 5-yr prospective study. Rheumatology (Oxford) 42, 778-783 (2003).
- Sokoll, K. B. & Helliwell, P. S. Comparison of disability and quality of life in rheumatoid and psoriatic arthritis. J. Rheumatol. 28, 1842-1846 (2001).
- Raychaudhuri, S. K. et al. Increased prevalence of the metabolic syndrome in patients with psoriatic arthritis.Metab. Syndr. Relat. Disord. 8, 331-334 (2010).
- Buckley, C. et al. Mortality in psoriatic arthritis—a single-center study from the UK. J. Rheumatol. 37, 2141-2144 (2010).
- Solomon, D. H. et al. Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis.Circulation 107, 1303-1307 (2003).
- Gladman, D. D. Mortality in psoriatic arthritis. Clin. Exp. Rheumatol. 26 (Suupl. 51), S62-S65 (2008) .
- Gladman, D. D., Farewell, V. T., Wong, K. & Husted, J. Mortality studies in psoriatic arthritis: results from a single outpatient center. II. Prognostic indicators for death. Arthritis Rheum. 41, 1103-1110 (1998).
- Jacobsson, L. T. et al. Treatment with TNF blockers and mortality risk in patients with rheumatoid arthritis. Ann. Rheum. Dis. 66, 670-675 (2007).
- Greenberg, J. D. et al. Tumour necrosis factor antagonist use and associated risk reduction of cardiovascular events among patients with rheumatoid arthritis. Ann. Rheum. Dis. 70, 576-582 (2011).
- Kramer, H. R. & Giles, J. T. Cardiovascular disease risk in rheumatoid arthritis: progress, debate, and opportunity. Arthritis Care Res. (Hoboken) 63, 484-499 (2011).
- Prodanovich, S. et al. Methotrexate reduces incidence of vascular diseases in veterans with psoriasis or rheumatoid arthritis. J. Am. Acad.Dermatol. 52, 262-267 (2005).
- Gupta, M. A. & Gupta, A. K. The Psoriasis Life Stress Inventory: a preliminary index of psoriasis-related stress.Acta Derm. Venereol. 75, 240-243 (1995).
- Schmid-Ott, G., Schallmayer, S. & Calliess, I. T. Quality of life in patients with psoriasis and psoriasis arthritis with a special focus on stigmatization experience. Clin. Dermatol. 25, 547-554 (2007).
- Salaffi, F., Carotti, M., Gasparini, S., Intorcia, M. & Grassi, W. The health-related quality of life in rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis: a comparison with a selected sample of healthy people.Health Qual. Life Outcomes 7, 25 (2009).
- Rapp, S. R., Feldman, S. R., Exum, M. L., Fleischer, A. B. Jr & Reboussin, D. M. Psoriasis causes as much disability as other major medical diseases. J. Am. Acad. Dermatol. 41, 401-407 (1999).
- Sprangers, M. A. et al. Which chronic conditions are associated with better or poorer quality of life? J. Clin. Epidemiol. 53, 895-907 (2000).
- Krueger, G. et al. The impact of psoriasis on quality of life: results of a 1998 National Psoriasis Foundation patient-membership survey. Arch. Dermatol. 137, 280-284 (2001).
- Fortune, D. G., Richards, H. L. & Griffiths, C. E. Psychologic factors in psoriasis: consequences, mechanisms, and interventions. Dermatol. Clin. 23, 681-694 (2005).
- Gupta, M. A., Schork, N. J., Gupta, A. K., Kirkby, S. & Ellis, C. N. Suicidal ideation in psoriasis. Int. J. Dermatol. 32, 188-190 (1993).
- Kavanaugh, A. F. & Ritchlin, C. T. Systematic review of treatments for psoriatic arthritis: an evidence based approach and basis for treatment guidelines. J. Rheumatol. 33, 1417-1421 (2006).
- Ritchlin, C. T. et al. Treatment recommendations for psoriatic arthritis. Ann. Rheum. Dis. 68, 1387-1394 (2009).
- Menter, A. et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 5. Guidelines of care for the treatment of psoriasis with phototherapy and photochemotherapy. J. Am. Acad. Dermatol. 62,114-135 (2010).
- Laine, L. Nonsteroidal anti-inflammatory drug gastropathy. Gastrointest. Endosc. Clin. N. Am. 6, 489-504 (1996).
- Gislason, G. H. et al. Increased mortality and cardiovascular morbidity associated with use of nonsteroidal anti-inflammatory drugs in chronic heart failure. Arch. Intern. Med. 169, 141-149 (2009).
- [No authors listed] Recommendations for use of selective and nonselective nonsteroidal antiinflammatory drugs: an American College of Rheumatology white paper. Arthritis Rheum. 59, 1058-1073 (2008).
- Dore, R. K. How to prevent glucocorticoid-induced osteoporosis. Cleve. Clin. J. Med. 77, 529-536 (2010).
- [No authors listed] Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis.ArthritisRheum. 44, 1496-1503 (2001).
- Black, R. L. et al. Methotrexate therapy in psoriatic arthritis; double-blind study on 21 patients. JAMA 189, 743-747 (1964).
- Willkens, R. F. et al. Randomized, double-blind, placebo controlled trial of low-dose pulse methotrexate in psoriatic arthritis. ArthritisRheum. 27, 376-381 (1984).
- Chandran, V., Schentag, C. T. & Gladman, D. D. Reappraisal of the effectiveness of methotrexate in psoriatic arthritis: results from a longitudinal observational cohort. J. Rheumatol. 35, 469-471 (2008).
- Lie, E. et al. Effectiveness and retention rates of methotrexate in psoriatic arthritis in comparison with methotrexate-treated patients with rheumatoid arthritis. Ann. Rheum. Dis. 69, 671-676 (2010).
- Menter, A. et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J. Am. Acad. Dermatol. 58, 826-850 (2008) .
- Kalb, R. E., Strober, B., Weinstein, G. & Lebwohl, M. Methotrexate and psoriasis: 2009 National Psoriasis Foundation Consensus Conference. J.Am. Acad. Dermatol. 60, 824-837 (2009).
- Pfizer. Azulfidine prescribing information [online], http://labeling.pfizer.com/ShowLabeling.aspx?id=524 (2009).
- Nash, P., Thagi, D., Behrens, F., Falk, F. & Kaltwasser, J. P. Leflunomide improves psoriasis in patients with psoriatic arthritis: an in-depth analysis of data from the TOPAS study. Dermatology 212, 238-249 (2006).
- Sanofi-Aventis. Arava prescribing information [online], http://products.sanofi.us/arava/arava.html (2010).
- Gottlieb, A. et al. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet 373, 633-640 (2009).
- Menter, A. et al. A randomized comparison of continuous vs. intermittent infliximab maintenance regimens over 1 year in the treatment of moderate-to-severe plaque psoriasis. J. Am. Acad. Dermatol. 56, 31.e1-31.e15 (2007).
- Pitarch, G., Sanchez-Carazo, J. L., Mahiques, L. & Oliver, V. Efficacy of etanercept in psoriatic patients previously treated with infliximab. Dermatology 216, 312-316 (2008).
- Haberhauer, G., Strehblow, C. & Fasching, P. Observational study of switching anti-TNF agents in ankylosing spondylitis and psoriatic arthritis versus rheumatoid arthritis. Wien. Med. Wochenschr. 160, 220-224 (2010).
- Haraoui, B. et al. Clinical outcomes of patients with rheumatoid arthritis after switching from infliximab to etanercept. J. Rheumatol. 31, 2356-2359 (2004).
- Sandborn, W. J. et al. Adalimumab induction therapy for Crohn disease previously treated with infliximab: a randomized trial. Ann. Intern.Med. 146, 829-838 (2007).
- Maini, R. N. et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 41,1552-1563 (1998).
- Atzeni, F. & Sarzi-Puttini, P. Autoantibody production in patients treated with anti-TNF-alpha. Expert Rev. Clin. Immunol. 4, 275-280 (2008).
- Abbott Laboratories. Humira prescribing information [online], http://www.rxabbott.com/pdf/humira.pdf (2011).
- Mease, P. J. et al. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double-blind, randomized, placebo-controlled trial. ArthritisRheum. 52, 3279-3289 (2005).
- Gladman, D. D. et al. Adalimumab for long-term treatment of psoriatic arthritis: forty-eight week data from the adalimumab effectiveness in psoriatic arthritis trial. Arthritis Rheum. 56, 476-488 (2007).
- Mease, P. J. et al. Adalimumab for long-term treatment of psoriatic arthritis: 2-year data from the Adalimumab Effectiveness in Psoriatic Arthritis Trial (ADEPT). Ann. Rheum. Dis. 68, 702-709 (2009).
- Amgen and Pfizer. Enbrel prescribing information [online], http://pi.amgen.com/united states/enbrel/derm/enbrel pi.pdf (2011).
- Mease, P. J. et al. Etanercept treatment of psoriatic arthritis: safety, efficacy, and effect on disease progression.Arthritis Rheum. 50, 2264-2272 (2004).
- Sterry, W. et al. Comparison of two etanercept regimens for treatment of psoriasis and psoriatic arthritis: PRESTA randomised double blind multicentre trial. BMJ 340, c147 (2010).
- Centocor Ortho Biotech. Remicade prescribing information [online],http://www.remicade.com/remicade/assets/hcp ppi.pdf (2011).
- Antoni, C. et al. Infliximab improves signs and symptoms of psoriatic arthritis: results of the IMPACT 2 trial. Ann. Rheum. Dis. 64, 1150-1157 (2005).
- Kavanaugh, A. et al. The Infliximab Multinational Psoriatic Arthritis Controlled Trial (IMPACT): results of radiographic analyses after 1 year. Ann. Rheum. Dis. 65, 1038-1043 (2006).
- van der Heijde, D. et al. Infliximab inhibits progression of radiographic damage in patients with active psoriatic arthritis through one year of treatment: Results from the induction and maintenance psoriatic arthritis clinical trial 2. Arthritis Rheum. 56, 2698-2707 (2007).
- Kavanaugh, A. et al. Golimumab, a new human tumor necrosis factor alpha antibody, administered every four weeks as a subcutaneous injection in psoriatic arthritis: Twenty-four-week efficacy and safety results of a randomized, placebo-controlled study. ArthritisRheum. 60, 976-986 (2009).
- Ko, J. M., Gottlieb, A. B. & Kerbleski, J. F Induction and exacerbation of psoriasis with TNF-blockade therapy: a review and analysis of 127 cases. J. Dermatolog. Treat. 20, 100-108 (2009).
- Centocor Ortho Biotech. Stelara prescribing information [online],http://www.stelarainfo.com/pdf/PrescribingInformation.pdf (2010).
- Mease, P. et al. Abatacept in the treatment of patients with psoriatic arthritis: results of a six-month, multicenter, randomized, double-blind, placebo-controlled, phase II trial. ArthritisRheum. 63, 939-948 (2011).
- Menter. A. et al. Adalimumab therapy for moderate to severe psoriasis: A randomized, controlled phase III trial.J.Am. Acad. Dermatol. 58, 106-115 (2008).
- Leonardi, C. L. et al. Etanercept as monotherapy in patients with psoriasis. N. Engl. J. Med. 349, 2014-2022 (2003).
- Gottlieb, A. B. et al. A randomized trial of etanercept as monotherapy for psoriasis. Arch.Derm. 139, 1627-1632 (2003).
- Chaudhari, U. et al. Efficacy and safety of infliximab monotherapy for plaque-type psoriasis: a randomised trial.Lancet 357, 1842-1847 (2001).
- Reich, K. et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet 366, 1367-1374 (2005).
- Gottlieb, A. B. et al. Infliximab induction therapy for patients with severe plaque-type psoriasis: a randomized, double-blind, placebo-controlled trial. J.Am. Acad. Dermatol. 51, 534-542 (2004).
- Antoni, C. E. et al. Sustained benefits of infliximab therapy for dermatologic and articular manifestations of psoriatic arthritis: results from the infliximab multinational psoriatic arthritis controlled trial (IMPACT). Arthritis Rheum. 52, 1227-1236 (2005).
http://www.medscape.org/viewarticle/749147_8
regards, taniafdi ^_^